
Moderna a breveté la séquence du gène du SRAS-CoV2 fabriqué en laboratoire militaire en 2018 !
Moderna a breveté la séquence du gène du SRAS-CoV2 fabriqué en laboratoire militaire en 2018 !
Extraits de l'article rédigé par Robert Bibeau et le Docteur Syed, publié le 24 janvier 2022 sur le site Les 7 du Québec


Nous le répétons inlassablement depuis presque deux ans, la pandémie de la COVID-19 a été transformé en exercice militaire viral mondial. Un virus (SRAS-CoV-2) s’est probablement échappé (ou s’est fait échappé!) du laboratoire de développement d’armes bactériologiques « à gain de fonction » de Wuhan et s’est répandu dans le monde entier. Les savants meurtriers, concepteurs de cette arme bactériologique associés aux multinationales pharmaceutiques (Big Pharma) n’avaient pas encore terminé de mettre au point le « vaccin » – traitement génique expérimental – pour contrer les effets les plus létales de cette arme virale démoniaque. Les firmes pharmaceutiques, en collusion avec les larbins politiciens mondiaux, ont tout de même décidé d’expérimenter ces traitements géniques sur la population mondiale de milliards de cobayes, faisant fi des dangers liés à cette expérience démente. L’article ci-dessous présente les preuves que le virus SRAS-CoV2, une arme virale et létale, concoctée dans les laboratoires de recherche militaires, étaient bien connues des trusts pharmaceutiques qui empochent les milliards de dollars au cours de ce qui est devenue une « plandémie » macabre. Il faut fermer ces laboratoires de recherche d’armes virales (une quarantaine dans le monde nous dit-on). Les preuves irréfutables de l’origine humaine du Covid-19 – les 7 du quebec Robert Bibeau.
« CTCCTCGGCGGGCACGTAG » est le pistolet fumant du Covid-19 qui fera la lumière sur cette pandémie.
Le génome du SRAS-CoV-2 contient une séquence inhabituelle de 19 nucléotides que l’on ne trouve nulle part ailleurs dans la nature – à part dans les brevets Moderna soumis en 2018.
SARS-CoV-2 genome contains a 19 nucleotide sequence #CTCCTCGGCGGGCACGTAG
probability of sequence occurring by chance: less than 1 in a billion
there’s no known virus including this sequence prior to SARS-Cov-2
Moderna has patents predating t pandemic that include this sequence
— bitbutter (@mormo_music) January 14, 2022

Selon les recherches approfondies menées par le Dr Ah Kahn Syed, Moderna a breveté la séquence unique trouvée dans le coronavirus deux ans avant le début de la pandémie, ce qui renforce la théorie selon laquelle le Covid-19 est un virus créé par l’homme.
Le Dr Syed écrit : Cela fait longtemps que je veux écrire cet article. Eh bien, au moins depuis que Prashant Pradhan (un merveilleux, honnête et courageux scientifique en génomique) a évoqué la possibilité, en février 2020, que le virus SARS-Cov2 ait été fabriqué par l’homme. Et nous avons vu de nombreuses pièces confirmant que le virus a été fabriqué dans un laboratoire, l’une des meilleures ici sur zenodo et avec sa propre vidéo ici. À l’heure où j’écris ces lignes, ces liens sont toujours en place, ce qui, après 12 mois, est une bonne chose pour tout article qui ose remettre en question le radotage propagé par notre chère « presse libre [sponsorisée par l’industrie pharmaceutique] ».
Quoi qu’il en soit, BLAST est le dépôt du NCBI/NIH (alias le gouvernement américain) pour les séquences génomiques et protéomiques, entre autres choses. C’est là que tous les généticiens du monde entier déposent leurs séquences lorsqu’ils font une découverte. Sa principale fonction est de permettre la comparaison des séquences de gènes et la découverte de séquences qui correspondent à une séquence que vous avez peut-être rencontrée dans votre expérience. Qu’est-ce qu’une séquence de gènes ? C’est simple. C’est une ligne de code, composée de n’importe quelle combinaison de 4 lettres dans une séquence. Vous vous souvenez du film GATTACA ? Si vous ne l’avez pas encore regardé, vous devriez, car il s’agit d’un autre film dystopique qui est maintenant trop proche de chez vous.
Le titre du film est basé sur les 4 bases nucléotidiques (G, A, T, C) qui constituent le code génétique de l’ADN de chaque être humain. Il y en a environ 3 milliards dans chaque cellule, ce qui donne un code unique – ce qui fait de vous un individu unique ! Le code s’apparie de telle sorte que G-C et A-T se combinent toujours pour former la double hélice que vous voyez sur l’image, par exemple GATTACA serait apparié à CTAATGT (le complément). Le code est lu dans un sens spécifique, de sorte que GATTACA sur un brin serait TGTAATC sur l’autre (le complément inverse). L’un des avantages de BLAST est qu’il ne se soucie pas de la version que vous lui donnez, il vous indiquera toujours le bon gène.
Une autre chose à noter à ce stade concerne les probabilités. Vous l’avez fait à l’école en lançant une pièce de monnaie (où le code est H pour pile ou T pour face). Quelle serait la probabilité de HHHHH (1 sur 2^4 = 1/16). Il en va de même pour TTTT. Il en va de même pour THTH, ou toute autre séquence spécifique de tirages à pile ou face. Essayez vous-même si vous ne me croyez pas (prédisez d’abord la séquence et voyez ensuite combien de fois vous devez l’exécuter). Le code génétique est essentiellement une « pièce de monnaie à quatre faces ». Ainsi, pour toute exécution d’une séquence spécifique (par exemple GATC), la probabilité d’obtenir cette séquence EXACTE est de 1 sur 4^4, ou pour tout nombre n de nucléotides (nt ou bases), la probabilité est de 1 sur 4^n (ceci est simplifié car dans certaines situations, la probabilité que la base suivante soit X dépend des bases environnantes).
BLAST a deux sections – nucléotide (BLASTn) et protéine (BLASTp). BLASTp traite les séquences d’acides aminés, de la même manière que les séquences de nucléotides. Mais il y a une grande différence car il y a 20 acides aminés (au lieu de 4 nucléotides) et par conséquent, même les séquences courtes (par exemple QTNS = Glu-Thr-Asn-Ser) auraient une probabilité d’environ 1 sur 20^4 (simplifié), soit 1 sur 160 000. La probabilité qu’une séquence spécifique de 5 acides aminés apparaisse par hasard sur la même base s’élève à 1 sur 3,2 millions !
Appuyons donc sur le bouton Protein BLAST et c’est parti… et voici l’écran que vous obtiendrez, que je vais vous faire parcourir.
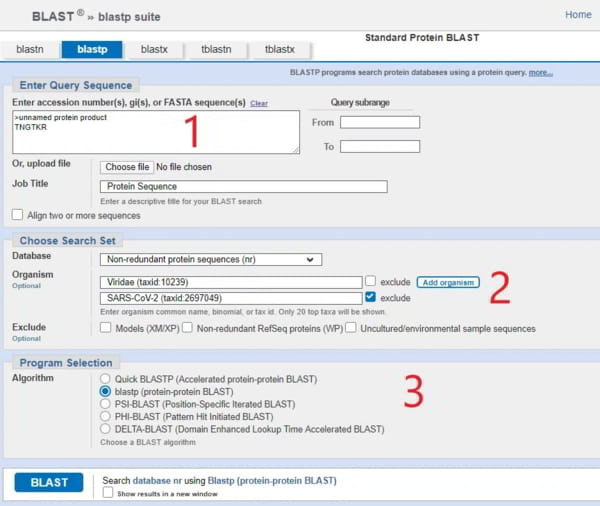
Dans [1], vous devez saisir la séquence d’acides aminés qui vous intéresse (BLAST ajoute automatiquement « >produit protéique sans nom »). Heureusement, vous n’avez pas besoin de chercher beaucoup car nous allons nous concentrer sur seulement 4 séquences dans le génome/protéome viral du SRAS-CoV-2 et celles-ci sont présentées dans le merveilleux article de Prashant Pradhan « Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag« publié le 31 janvier 2020 quelques jours après la publication de la séquence du génome.
La partie dont vous avez besoin se trouve dans le tableau 1 que je publie ici et vous verrez que j’ai affiché la séquence de 6 acides aminés TNGTKR dans la case [1] marquée en rouge sur l’écran BLASTp.
Pour l’étape [2] de l’écran de saisie de BLASTp, vous devez ajouter quelques filtres. Le premier filtre consiste à restreindre la recherche aux « viridae (virus) » (ou vous pouvez simplement entrer 10239 qui est l’ID taxonomique). La raison en est qu’il existe des milliards d’espèces sur la planète et que BLASTp les recherchera toutes, mais que vous voulez seulement savoir de quel virus provient ce motif. Il n’est pas vraiment intéressant de savoir si le motif est trouvé dans un calmar, bien qu’il soit possible qu’un calmar ait également participé à l’action avec la fameuse chauve-souris et le pangolin, dont parlent des gens comme Peter Daszak et Dominic Dwyer, ce qui en fait une ménagerie zoonotique, mais restons dans la réalité.
La deuxième condition est d’exclure toutes les références au SRAS-CoV-2 qui se sont maintenant accumulées dans la base de données, car elles vont toutes apparaître (des milliers) et cela ne nous intéresse pas.
Une fois que vous avez saisi ces données, appuyez sur le bouton BLAST et qu’obtenez-vous ? Vous obtiendrez une liste de candidats qui ont une homologie proche de cette séquence. Comme il s’agit d’une séquence très courte, l’homologie (ressemblance) devrait être de 100 %. Le haut de la page est un résumé de ce que vous avez demandé et le reste de la page est une liste des séquences correspondantes. Ce que vous verrez immédiatement, c’est qu’en haut de la liste se trouvent deux virus synthétiques qui sont une chimère du SRAS-Cov-2 et un autre virus, apparu au cours des deux dernières années grâce à des laboratoires qui fabriquent d’autres virus (parce que nous n’en avons pas assez). Le suivant dans la liste est un tas de références au VIH-1.

Vous pouvez cliquer sur n’importe lequel d’entre eux et vous serez amené à l’écran d’alignement où les alignements entre le sujet (votre TNGTKR) et la requête (tous les virus) sont affichés, et vous verrez en descendant la page que les alignements ne tiennent que pour le VIH-1 jusqu’à ce que vous commenciez à obtenir des protéines synthétiques et hypothétiques, jusqu’au prochain vrai virus dans la liste qui est le VIH-2….
OK, mais un résultat comme celui-ci pourrait être une coïncidence. Dans la liste, vous verrez la « valeur E » qui est un indicateur de la probabilité de trouver des correspondances comme celle-ci et qui doit être aussi proche de zéro que possible. Ici, elle est de 282, ce qui ne fait que refléter la probabilité de trouver des correspondances sur une séquence courte.
Donc, voilà le problème. Soit ces séquences vont par hasard correspondre à tout un tas d’autres virus (parce que la valeur E est élevée et que nous devons donc nous attendre à de nombreuses correspondances), soit il s’agit de séquences vraiment inhabituelles qui ont spécifiquement, préférentiellement ou uniquement correspondu au VIH-1. Comment aborder cette question ? Eh bien, passons à la séquence suivante – HKNNKS, qui est une autre séquence courte. Pour mettre de l’ordre dans l’écran, nous pouvons définir un filtre pour les séquences courtes afin de nous assurer que 100% de la séquence correspond (en haut à droite). Maintenant, souvenez-vous que si la correspondance avec le VIH-1 était aléatoire, nous ne devrions pas vraiment voir de correspondance sur cette liste, car elle devrait être éliminée par toutes les autres centaines de virus qui devraient correspondre de manière préférentielle. Oups…
